Механизмы человеческих заболеваний, вызываемых прионами (англ. от Protein («белок») и Infection («Инфекция»); впервые слово было использовано С. Прузинером [1] в конце XX века), до сих пор остаются малоизученными, несмотря на, казалось бы, большой объём исследований, проводимых в данной сфере. Цель настоящей статьи заключается в обобщении и понятном объяснении имеющейся на данной момент информации, касающейся прионов и ассоциированных с ними болезней.
В рамках настоящей статьи приняты следующие сокращения: prion — proteinacious infectious particle; PrP — прионный белок; PrPC — нормальная изоформа прионного белка; PrPSc — инфекционная форма прионного белка; PRNP — ген, кодирующий прионный белок.
Прионные заболевания (также известны как трансмиссивные губчатые энцефалопатии, ТГЭ), ставшие известными человеку в середине XVIII века, являются одним из наиболее интригующих биологических феноменов. Исследования этого явления начались в XX веке с попыток определить биологическую сущность возбудителей сразу нескольких специфичных болезней животных и человека со схожей симптоматикой. Гипотеза об их общей этиологии, выдвинутая в 1960-х годах учёными радиобиологом Т. Альпером и математиком Д. Гриффитом [2] и позже дополненная и доказанная врачом С. Прузинером [3], дала толчок последующим исследованиям в этой области. Однако, несмотря на глубокую заинтересованность научного мира, многие аспекты существования прионов остаются неизученными и по сей день.
К числу человеческих болезней, связанных с этими особыми белковыми инфекционными агентами, относятся болезнь Якоба-Крейтцфельда (CJD) и её различные вариации, фатальная бессоница (FFI/FSI), болезнь Герстмана-Шраусслера-Шейнкера (GSS), куру, вариабельная протеаза-чувствительная прионопатия, и прионное заболевание, связанное с диареей и поражением вегетативной нервной системы.
Все вышеперечисленные прионные болезни на сегодняшний день остаются смертельными, что помещает их в категорию наиболее опасных болезней.
Сущность прионов
После окончания процесса перевода генетической информации, заключённой в виде нуклеотидной последовательности РНК (рибонуклеиновой кислоты), в специфическую последовательность аминокислот, формирующую первичную структуру всех протеинов, новосинтезированные белки сворачиваются в определённые структуры. Прионы — разновидность белковых молекул с неправильной «укладкой» (Рис. 1), дефектная форма нормального мембранного белка PrP, который экспрессируется (проявляется) преимущественно в клетках центральной нервной системы.
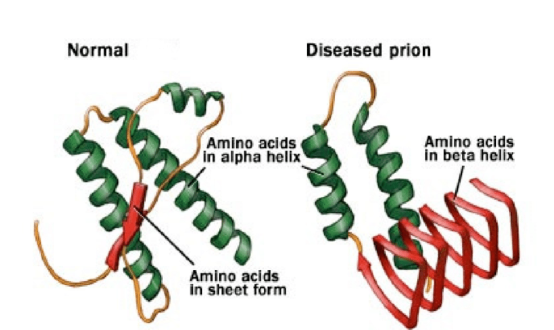
Рис. 1. Правильно «сложенный» (слева) и дефектный (справа) белки [4]
Благодаря определённым биологическим машинериям, обычные неправильно сложенные белки легко утилизируются и не оказывают отрицательного эффекта на процессы человеческой жизнедеятельности. Прионы отличаются устойчивостью к этим механизмам и наличием способности превращать нормальные составляющие их белки в себе подобные. Существует две гипотезы, описывающие механизмы этого явления. Согласно первой, гетеродимерной модели (Рис. 2) [5], превращение происходит следующим образом: PrPSc присоединяется к «здоровой» молекуле PrP, и катализирует ряд конформационных изменений, приводящих к её переходу в прионную форму, после чего уже два ненормальных белка расходятся и запускают новые раунды этого процесса. При этом наличие агрегированной («склеенной») формы белка не является обязательной частью прионной трансформации.
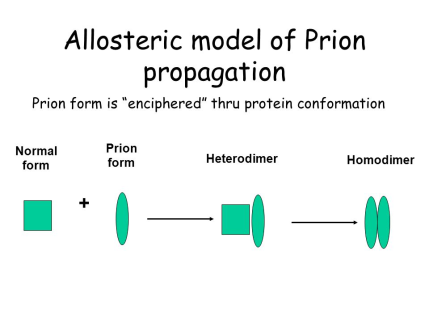
Рис. 2. Гетеродимерная модель прионной репликации [6]
Альтернативная гипотеза (Рис. 3) [5] — полимеризационная — гласит, что катализ конформационного превращения нормального белка в патологический может происходить только при «нуклеализации» с последующим образование олигомерных или мультимерных комплексов. Стоит отметить, что последние исследования говорят в пользу второй модели.
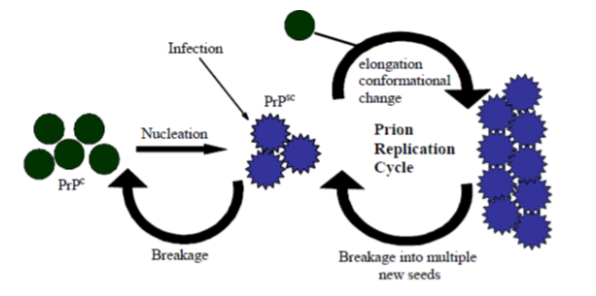
Рис. 3. Цикл репликации PrPSc в соответствии с полимеризационной гипотезой [7]
Воздействие PrPSc приводит к этакой «внутриклеточной эпидемии»: образуется множество нефункциональных белковых бляшек на клетках организма, из-за чего он рано или поздно погибает.
Пути возникновения прионных заболеваний
Считается, что существует всего три пути приобретения прионных заболеваний: прямое инфицирование, наследственная передача и спорадическое возникновение по неизвестному механизму [8], но вне зависимости от происхождения они могут быть переданы инфекционным путём.
Был детектирован высоконсервативный ген PRNP, несущий информацию о нормальной изоформе белка PrP, находящийся в p-плече 20-ой хромосомы человека [9]. PRNP имеет протяженность 16 тысяч нуклеотидных последовательностей и содержит 2 экзона. Все наследственные прионные заболевания связаны с аутосомным наследованием мутаций, произошедших в данном гене.
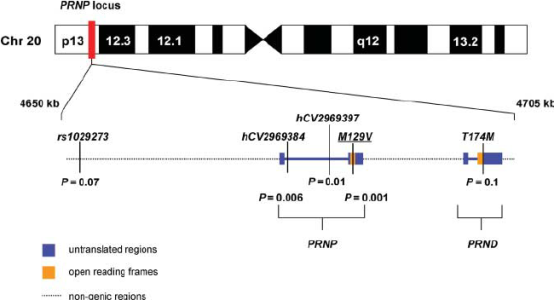
Рис. 4. Локализация гена PRNP в 20-ой хромосоме человека [10].
Основной способ возникновения прионной болезни — спонтанный. Согласно одной из гипотез, объясняющих этот процесс, в нормальных белках происходит определённая посстрансляционная модификация [11]. Иная гипотеза постулирует, что в какой-то конкретный момент неопределённое количество клеток организма соматически (ненаследственно) мутирует и начинает производить дефектный белок PrPSc [12].
Попасть в незаражённый человеческий организм прион может различными путями: при поедании плохоприготовленного мяса, содержащего PrPSc, переливании крови от инфицированного человека к здоровому, трансплантации заражённых органов и тканей.
Клиническая картина прионных заболеваний человека
Ещё одной важной особенностью прионных белков является наличие возможности принимать определённое число различных конформаций. Это обуславливает отличия в течении и симптоматике прионных заболеваний: возможны различные инкубационные периоды, повреждения разных участков коры головного мозга, нарушения различных функций нервной системы [13]. Несмотря на это, среди всех ассоциированных с действием прионов заболеваний прослеживается серия общих чёрт: поражение нервной системы, изначальное отсутствие иммунного ответа на дефектные белки PrP вследствие постоянного присутствия их «правильной» изоформы в организме [5], быстрая прогрессия болезни после окончания инкубационного периода.
Болезнь Крейтцфельда-Якоба (CJD)— редкое, но тем не менее наиболее известное прионное заболевание человека. Существует несколько форм CJD — наследственная, ятрогенная, спорадическая, вариативная, при этом первые три отличаются преимущественно способом распространения. Патоморфологические картины наследственной, ятрогенной и спорадической CJD схожи: во всех случаях наблюдаются прогрессирующие когнитивные нарушения, поражения и дисфункуция мозжечка или комбинация этих расстройств; нарушение зрения вплоть до слепоты; миоклонические припадки. В терминальной стадии появляются глобальные когнитивные нарушения, смерть наступает через 8–10 месяцев после диагностирования CJD [14]. Вариативная CJD имеет несколько более глубоких отличительных особенностей: поражает молодых людей в возрасте в среднем до 30 лет, ее начало характеризуется изменениями поведения, бессонницей, депрессией; двигательные нарушения проявляются примерно через 6 месяцев от начала заболевания в виде прогрессирующей атаксии, хореи, миоклонуса; слабоумие наступает позднее, чем при классической форме, пациент осознает свое ухудшающееся состояние. Для вариабельной БКЯ типичны не только начало в более молодом возрасте, но и средняя выживаемость, превышающая 14 месяцев [15].
Болезнь Герстмана-Штраусслера-Шейнкера (GSS)— заболевание, несколько отличающееся от CJD по нескольким признакам. Эта клиническая форма ТГЭ вызвана мутацией гена PNRP в 102-ом кодоне, приводящей к замене аминокислоты пролин на лейцин [16]. Болезнь начинается в среднем возрасте с проявления мозжечковой атаксии, речевых расстройств, деменции и измнений в поведении. К числу этих симптомов могут прибавляться диплопия, глухота, миоклонические приступы, спастичность.
Куру— прионное заболевание, эндемичное для некоторых районов Папуа-Новой Гвинеи. Основным способом распространения этой болезни был ритуальный каннибализм. Симптоматика включает в себя двигательные расстройства (тремор, массивные фасцикуляции, хореоатетоз, миоклонии). Смерть наступает через приблизительно 2 года после дебюта заболевания. В 2009 году было уставлено, что некоторые члены одного из аборигенных племён обладают врождённым иммунитетом к куру благодаря появлению у них относительно новой полиморфической модификации гена PNRP [17].
Фатальная бессоница — редкое ТГЭ, обычно связанная с наследованием аутосомно-доминантной мутации («семейная», FFI). Отмечается, что существует и спорадическая форма этого заболевания («спонтанная», FSI) [18]. В обоих случаях наблюдается следующая картина: нарушения сна, галлюцинации, вегетативная гиперактивация, двигательные нарушения, резкое и прогрессирующее снижение когнитивных способностей. Болезнь продолжается от 8 до 72 месяцев (среднее значение составляет около 18 месяцев), после чего больной умирает. Происходит стремительная гибель нейронов и астроглиоз переднего и медиального таламуса и нижних оливок с последующим поражением коры головного мозга и мозжечка. Описана стадийность заболевания [18]: начальная форма характеризуется появлением тяжёлой бессонницы, панических атак, патологической тревоги и фобий; на второй стадии развития болезни больной начинает испытывать галлюцинации; на предпоследней стадии больной утрачивает способностью ко сну и стремительно теряет вес; терминальная стадия приводит к утрате речи и смерти.
В 2013 году была обнаружена иная клиническая форма прионных заболеваний, связанная с поражением вегетативной нервной системы [19]. Заболевание ассоциировано с появлением новой мутации в гене PRNP, приводящей к укорочению прионного белка и последующим нарушением его связи с клеточными мембранами. В этом случае распространение прионных агрегатов не ограничивается ЦНС. Предположительно, имеет место миграция PrPSc в переферические нервы и внутренние органы. Серия симптомов проявляется уже в раннем возрасте: хронический понос, вегетативная недостаточность и сенсорная полинейропатия. В зрелом возрасте наступает поражение ЦНС, приводящее к появлению деменции и судорог в числе симптомов.
Вариабельная протеаза-чувствительная прионопатия (VPSPr, ВПЧП) — ещё одно новое редкое спорадическое прионное заболевание, впервые описанное в 2008 году. VPSPr схожа с GSS по особенностям PrPSc, однако в прионе VPSPr не было детектировано каких-либо мутаций гена PRNP [20]. Невосприимчивость прионных белков, вызывающих VPSPr, к действию протеаз, значительно снижена. Болезнь манифестируется расстройствами речи (афазия, дизартрия), когнитивными нарушениями, в некоторых случаях атаксией и паркинсонизмом.
Перспективы лечения прионных заболеваний
Множество исследований в этой области дают основание для возможности ингибиции репликации прионов и лечения вызывамых ими болезней.
Как было отмечено выше, иммунный ответ организма на PrPSc отсутствует. Однако эксперимент, проведённый с полученными in vitro прионами, доказал, что использование антител, выработанных на определённые антигенные детерминанты PrP, индуцирует ингибирование размножения PrPSc [21], приводящее к отсрочке заболевания. Прионное превращение также может быть остановлено с помощью «блокаторов β-структур» — пептидных последовательностей, обогащенных аминокислотой пролином и обладающих гомологичным PrPC составом [5]. Иной подход основан на использовании антисенсовых олигонуклеотидов (АСО) — коротких фрагментов нуклеиновых кислот, останавливающих трансляцию с матричной РНК за счёт образования на ней петлеобразных участков. Именно АСО на данный момент является наиболее эффективным методом ингибиции репликации прионов: эксперименты с введением АСО в спинномозговую жидкость лабораторных мышей, проведённые в Лабораториях Скалистых Гор (Rocky Mountain Laboratories), привели к отсрочке проявления прионных болезней у подопытных на 113–135 дней [22]. Астемизол, относящийся к группе блокаторов H1-гистаминовых рецепторов (H1R), обладает доказанным антиприонным действием [23]. Кроме этого, для предотвращения размножения прионов возможно использование мутаций гена PNRP, приводящих к изменениям Q171R и E219K в аминокислотной последовательности PrP: мутантные прионные белки неспособны переходить в патологическую форму [24].
В данный момент лечение прионных заболеваний может быть только симптоматическим. Использование Брефельдина А, разрущающего аппарат Гольджи и тем самым замедляющего процесс распространения PrPSc и антагонистов NMDA-рецепторов, способствующих более длительному выживанию инфицированных клеток, в терапии CJD не достигло большого успеха [25]. Попытки применения классических противовирусных средств в лечении CJD и GSS также оказались неудачными [25]. Традиционные снотворные обладают нулевой эффективностью в терапии FSI и FFI, хотя был зафиксирован случай отсрочки летального исхода при одновременном использовании ряда сильнодействующих препаратов (диазепама, кетамина, оксида азота) [26]. Терапия VPSPr может быть основана на использовании повышенной чувствительности PrPSc к действию протеаз: доставка в организм смеси специфических иммобилизованных цистеиновых протеаз, вероятно, позволит отсрочить проявление болезни и пролонгировать жизнь больного. Этиотропное лечение основных ТГЭ, судя по всему, должно быть основано на использовании вышеперечисленных или подобных им методов ингибиции прионной репликации.
Литература:
- Stanley B. Prusiner — Autobiography. NobelPrize.org.
- Alper T., Cramp W. A., Haig D. A., Clarke M. C. Does the agent of scrapie replicate without nucleic acid? // Nature. — 1967. — May (vol. 214, no. 5090). — P. 764–766.
- Taubes, Gary. The game of name is fame. But is it science? // Discover. — 1986. — December (т. 7, № 12). — С. 28–41.
- Mayo Foundation of Medical Education and Research.
- И. С. Шкундина, М. Д. Тер-Аванесян. Прионы. Успехи биологической химии, т. 46, 2006. — С. 7–9.
- Nora Whisler. Lecture 34: PRION. — 2015. — P. 22.
- Dipendra Paj Pandeya, Nimish K. Acharya and Seong-Tshool Hong. Review: The Prion and its Potentiality. // Biomedical Research. — 2010.
- Groschup M. H., Kretzschmar H. A., eds. Prion Diseases Diagnosis and Pathogeneis // Archives of Virology — New York: Springer, 2001. — Vol. Suppl 16.
- Oesch B., Westaway D., Wälchli M., et al. A cellular gene encodes scrapie PrP 27–30 protein // Cell. — Cell Press, 1985. — Vol. 40, no. 4. — P. 735–746.
- Andreas Papassotiropoulus, Adriano Aguzzi, M. Axel Wollmer, Christoph Hock. The prion gene is associated with human long-term memory. // Human Molecular Genetics. — 2005.
- Prion Diseases (Transmissible Spongiform Encephalopathies) [Online].
- Prion Clinic: Sporadic Prion Disease [Online].
- Н. Н. Заваденко, Г. Ш. Хондкарян, Р. Ц. Бамбеева, А. А. Холин, Е. Н. Саверская. Прионные заболевания человека: современные аспекты. // Журнал неврологии и психиатрии им. С. С. Корсакова. — 2018. — т. 118, изд. 6. — C. 91–92.
- ICTVdB Index of Viruses. U. S. National Institutes of Health website [Online].
- Clinical and Pathologic Characteristics | Variant Creutzfeldt-Jakob Disease, Classic (CJD) [Online].
- Arata H, Takashima H, Hirano R, et al. Early clinical signs and imaging findings in Gerstmann–Sträussler–Scheinker syndrome (Pro102Leu). // Neurology. — 2006.
- Mead S, Whitfield J, Poulter M, Shah P, Uphill J, Campbell T, Al-Dujaily H, Hummerich H, Beck J, Mein CA, Verzilli C, Whittaker J, Alpers MP, Collinge J. A Novel Protective Prion Protein Variant that Colocalizes with Kuru Exposure. // New England Journal of Medicine. — 2009.
- R. Turner. Dying To Sleep: Fatal Familial Insomnia (FFI) [Online].
- Mead S, Gandhi S, Collinge J, Caine D, Gallujipali D, Carswell Ch, Hyare H, Joiner S, Ayling H, Lashley T, Linehan JM, Al-Doujaily H, Sharps B, Revesz T, Sandberg MK, Reilly MM, Koltzenburg M, Forbes A, Rudge P, Brandner S, Warren JD, Wadsworth JDF, Wood NW, Holton JL, Collinge J. A novel prion disease associated with diarrhea and autonomic neuropathy. // New England Journal of Medicine. — 2013.
- Gambetti P, Puoti G, Zou WQ. Variably protease-sensitive prionopathy: a novel disease of the prion protein. // Journal of Molecular Neuroscience. — 2011.
- M. Enari, E. Flechsig, C. Weissmann. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. // Proceedings of the National Academy of Sciences. — 2001.
- G. Raymond, et al. Antisense oligonucleotides extend survival of prion-infected mice. // JCI Insight. — 2019.
- Scripps Research Institute Scientists Identify First Potentially Effective Therapy for Human Prion Disease; Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents // Proceedings of the National Academy of Sciences. — 2013.
- K. Kaneko et al. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. // Proceedings of the National Academy of Sciences. — 1997.
- M. Maniz, P. Kalakoti, M. Henry, J. Thakur, R. Menger, B. Guthikonda, A. Nanda. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. // Neurosurgical Focus. — 2015.
- Дэвид Робсон. Трагедия людей, переставших спать: можно ли им помочь? [Online] // BBC Future. — 2016.

