Кристаллическая дистрофия Биетти (КДБ) — редкое аутосомно-наследственное наследственное заболевание, вызываемое мутациями в CYP4V2 гена. Профессор Джан Баттиста Битетти впервые описал это заболевание в 1037 году [1], сообщив о трех пациентах, включая двух братьев, с кристаллическими пятнами сетчатки в заднем полюсе, рассеянными скоплениями пигмента сетчатки, атрофией и поверхностными отложениями в роговице.
КДБ наследуется по аутосомно-рецессивному типу, что означает, что обе копии гена в каждой клетке имеют мутации. Каждый из родителей человека с аутосомно-рецессивным заболеванием- несут по одной копии мутировавшего гена, но обычно не проявляют признаков и симптомов этого состояния. [7] КДБ связана с мутацией гена CYP4V2, этот ген способствует созданию фермента цитохрома P450, эти ферменты участвуют в образовании и распаде различных химических веществ в клетках, фермент CYP4V2 участвует в многоступенчатом процессе, называемом окислением жирных кислот, в котором липиды расщепляться и превращаются в энергию, при мутациях гена CYP4V2 функция фермента нарушается, и это влияет на распад липидов.
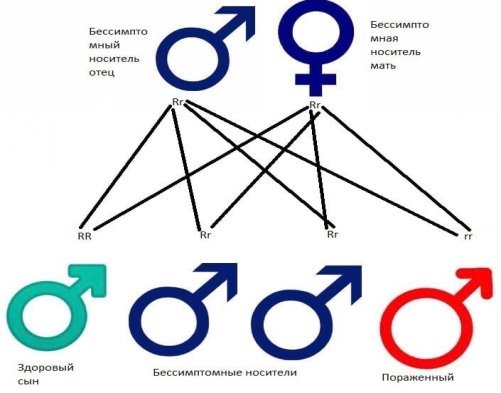
Рис.1 Рецессивный образец наследования
Оценить распространенность КДБ довольно сложно, так методы сбора информации отличаются, в зависимости от страны, авторов и опросов, автор Hu [2], сделал заключение и оценил частоту гена 0,005, изучая первых двоюродных братьев и сестер при обследовании в Китае в 1983 году. Автор Okialda et al [3] оценил распространенность КДБ на 1 человека из 67000 человек, из которых страдают 21 000 пациента в Китае и около 5 000 в США. Но данное заболевание распространенно по всему миру и является обычным явлением на востоке Азии, являясь более распространенным среди китайского, японского и корейского населения, так же много есть зарегистрированных случаев, диагностированных в Европе, относящиеся к пациентам из Италии, Ливана и Испании, вследствии чего можно сделать вывод, что возможно и средиземноморское распространение болезни [4].
КДБ — это прогрессирующая дистрофия с поражением роговицы, характеризующаяся обильными желтоватыми сверкающими отложениями на сетчатке, потерей хориокапилляров, атрофией и кристаллических отложений в периферической роговице.
Кристаллическую дистрофию Биетти Yuzawa et al [5], разделил на 3 стадии:
1 стадия — атрофия пигментного эпителия сетчатки с кристаллическими отложениями в области макулы;
2 стадия — атрофия пигментного эпителия сетчатки выходит за пределы заднего полюса, происходит атрофия хорикокапилляров на заднем полюсе;
3 стадия — обширная атрофия пигментного эпителия сетчатки и хорикокапилляров, кристаллические отложения по всему глазному дну.
Первые клинические проявления будут проявляться между вторым и третьим десятилетием жизни, но могут развиваться у подростков и у пожилых людей, самые ранние стадии протекают бессимптомно, что усложняет его диагностику. Поэтому при диагностировании заболевания на самых ранних стадиях происходит часто случайным образом, по мере прогрессирования КДБ симптомы будут проявляться медленно и безболезненно, ограничивая периферическое зрение и сужение поля зрения, нарушение цветового зрения, помутнения, фотопсия. На поздних стадиях заболевание обнаруживается уже с серьезными нарушениями зрения. Так же стоит подметить, что заболевание может быть асимметричным в обоих глазах. [6].
Диагностика КДБ основана на офтальмологической оценке многочисленных мелких блестящих желто-белых кристаллических отложений в сетчатке или же без каких-либо кристаллических отложений в роговице, атрофии пигментного эпителия сетчатки и склероза сосудистой оболочки глаз, [8] кристаллы обычно изолированы в субэпителиальной и передней строме периферической роговицы [9]. Диагностические инструменты включают электроретинографию для оценки степени дисфункции палочек и колбочек, тест поля зрения Хамфри для выявления дефицита поля зрения, зеркальная микроскопия [10]. Так же можно прибегнуть к молекулярно-генетическому тестированию для идентификации патологических вариантов CYP4V2, если клинические признаки сомнительны [11].
Литература:
- Bietti G. Ueber faxmiliares Vorkommen von «Retinitis punctata albescens» (verbunden mit «маргинальная дистрофия роговицы»), glitzern, des glaskorpers und anderen degenerativen augenveranderungen. Klin Monbl Augenheilkd. 1937; 99: 737–756.
- Hu DN. Ophthalmic genetics in China. Ophthal Paed Genet. 1983;2:39–45.
- Okialda KA, Stover NB, Weleber RG et al. Bietti Crystalline Dystrophy. In: Pagon RA, Bird TD, Dolan CR et al. editors. Gene ReviewsTM. Seattle (WA) University of Washington, Seattle: 1993–2014
- García-García GP, Martínez-Rubio M, Moya-Moya MA, Pérez-Santonja JJ, Escribano J. Identification of novel CYP4V2 genotypes associated with Bietti crystalline dystrophy and atypical anterior segment phenotypes in Spanish patients. Acta Ophthalmol. 2018;96(7):e865–e873. doi:10.1111/aos.13768
- 5-Yuzawa M, Mae Y, Matsui M. Bietti’s crystalline retinopathy. Ophthalmic Paediatr Genet. 1986;7:9–20. doi:10.3109/13816818609058037
- Haddad NM, Waked N, Bejjani R, et al. Clinical and molecular findings in three Lebanese families with Bietti crystalline dystrophy: report on a novel mutation. Mol Vis. 2012;18:1182–1188
- Li, A; Jiao, X; Munier, Fl; Schorderet, Df; Yao, W; Iwata, F; Hayakawa, M; Kanai, A; Shy, Chen, M; Alan, Lewis, R; Heckenlively, J; Weleber, Rg; Traboulsi, Ei; Zhang, Q; Xiao, X; Kaiser-Kupfer, M; Sergeev, Yv; Hejtmancik, Jf. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2 (англ.) // American Journal of Human Genetics (англ.): journal. — 2004. — May (vol. 74, no. 5). — P. 817–826. — doi:10.1086/383228. — PMID 15042513.
- Furusato E, Cameron JD, Chan CC. Evolution of Cellular Inclusions in Bietti's Crystalline Dystrophy. Ophthalmol Eye Dis. 2010;2010(2):9–15.
- Krachmer J, Mannis M, Holland E: CORNEA, 4th ed.Elsevier Mosby, 2017, 251–264.
- Vargas M, Mitchell A, Yang P, Weleber R. Bietti Crystalline Dystrophy. GeneReviews. 2012.
- Xiao X, Mai G, Li S, Guo X, Zhang Q. Identification of CYP4V2 mutation in 21 families and overview of mutation spectrum in Bietti crystalline corneoretinal dystrophy. Biochem Biophys Res Commun. 2011;409(2):181–6.

