Работа посвящена изучению прогноза метаболизма лекарственных взаимодействий in vivo из данных in vitro. Согласно исследованию, это хорошая экономия времени и стоимости в лекарственных разработках. Тем не менее, in vivo-in vitro экстраполяция лекарственных взаимодействий является сложной и спорной. Общий подход: использовать значения Кi и Кm in vitro вместе с плазменной концентрацией ингибиторов in vivo, чтобы прогнозировать возможности лекарственных взаимодействий in vivo.
Ключевые слова: фармакокинетика, компьютерная симуляция, метаболизм лекарств.
Введение. Фармакокинетическое взаимодействие лекарственных средств может приводить к серьезным неблагоприятным явлениям, и оценка возможных взаимодействий между ними — составная часть развития лекарств и норматив их утверждения на рынке. Оптимальные рекомендации по приему лекарств в определенном режиме для пациентов, которым назначено много препаратов одновременно, не только повышают эффективность лечения, но и позволяют снизить нежелательные побочные реакции организма в ответ на терапию. Желательные и нежелательные эффекты лекарственных средств обычно связаны с и концентрацией в месте действия, что, в свою очередь, связано с количеством вводимого вещества (дозы) и его абсорбцией, распределением, метаболизмом и /или экскрецией. На все эти процессы могут влиять как внутренние, так и внешние факторы, такие как возраст, раса, пол, болезни, принимаемые лекарства, пища, соки [2]. Наблюдаемые изменения могут быть существенными, например, на порядок или более увеличение или уменьшение концентрации препарата или его метаболитов в крови и тканях. Многие из этих взаимодействий участвуют в ингибировании метаболизирующих ферментов и переносчиков, что приводит к увеличению системного воздействия и последующим побочным реакциям [3]. Изменение активности фермента и /или переносчика, участвующего в абсорбции, распределении, метаболизме или экскреции нового молекулярного объекта, другими сопутствующими препаратами, может привести к изменению воздействия, приводящему к изменению реакции (безопасности или эффективности). На протяжении многих лет были разработаны различные методы изучения in vitro для прогнозирования взаимодействия лекарственных средств между собой in vivo. Исследование in vitro стало важнейшим шагом в оценке взаимодействия лекарственных средств. Хорошо выполненные исследования in vitro могут быть использованы в качестве скрининга для дальнейшей оценки in vivo и могут служить основой для разработки последующих исследований взаимодействия лекарств in vivo. Кроме экспериментов in vitro, компьютерное моделирование и симуляция также могут помочь в прогнозировании взаимодействия лекарственных средств [6].
Цель. Составить оптимальные рекомендации по приему лекарств в определенном режиме для пациентов, которым назначено много препаратов одновременно.
Материалы и методы. Проведен систематический поиск информации о моделях лекарственных взаимодействий в медицинских базах данных Pubmed, ссылках медицинских публикаций. Текущая модель поясняет растущую концентрацию лекарства в печени при использовании лекарственных концентраций в вене портае вместо системного кровотока и избегает использования сомнительного коэффициента распределения печень/плазма, чтобы компенсировать снижение ожидаемой ингибирующей концентрации [1].
Модель основывается на следующем:
1. Метаболическое ингибирование — это только механизм взаимодействия между двумя лекарствами.
2. Ингибирование метаболизма субстрата обратимо и подчиняется закону Михаэлиса.
3. Концентрация лекарства в портальной вене была такой же, как концентрация энзимов в печени.
4. Сpv использовалась как концентрация лекарства по уравнению Михаэлиса.
5. В модели не рассматривается количество вещества, связанного белками плазмы.
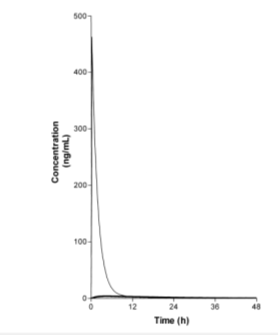
Рис. 1. Смоделированые результаты концентрации лекарства, принятого p. o., в системном кровотоке (жирная линия) и в портальной вене (тонкая линия)*.
*Исходные параметры модели: ka=0.01 мин-1,Vd= 20000 мл/кг, Eh=0.5
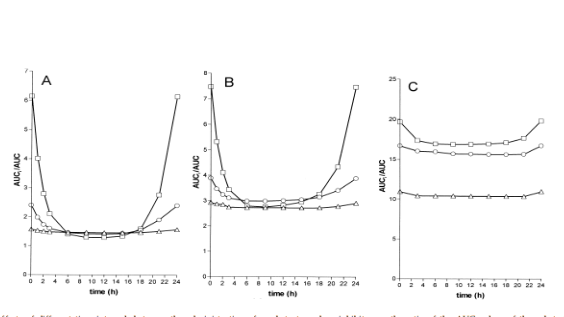
Рис. 2. Эффекты, наблюдаемые при различных временных интервалах назначения субстрата и ингибитора, в соотношении со значением AUC субстрата к AUC ингибитора.*
*Eh ингибитора = 0.9 (А), 0.5 (В), 0.1 (С). Еh субстрата =0.9 (квадрат), 0.5 (круг), 0.1 (треугольник), ka=0.01 мин-1; Vd= 20000 мл/кг
Результаты исследования. Управляя субстратом и ингибитором в различное время, количество нежелательных лекарственных взаимодействий возможно снизить. На этой стратегии должен основываться режим дозирования препаратов у пациентов, которым назначено несколько лекарственных средств одновременно.
Выводы.
- Концентрация лекарства в портальной вене намного выше, чем в системном кровотоке в фазу абсорбции, и должна быть использована для прогнозирования метаболического ингибирования лекарственных взаимодействий.
- Самое высокое ингибирование наблюдалось, когда два лекарства назначались в одно и то же время.
- При запаздывании назначении ингибитора, следующего за субстратом, возможно значительно снизить показатель лекарственного взаимодействия.
Литература:
- Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. Prediction of pharmacokinetic alterations caused by drug-drug interaction: metabolic interaction in the liver. Parmacol Rev 1998; 50: 387–411.
- Huang SM, Temple R. Is this the drug or dose for you? Impact and consideration of ethnic factors in global drug development, regulatory review, and clinical practice. Clin Pharmacol Ther 2008:84(3):287–4.
- In vivo drug metabolism/drug interaction studies-study design, data analysis, and recommendations for dosing and labeling. http://www.fda.gov/cder/guidance.
- Lin JH, Lu AYH. Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet 1998; 35: 361–390.
- Bergstorm RF, Peyton AL, Lemberger L. Quantification and mechanism of the fluoxetine and tricyclic antidepressant interaction. Clin Pharmacol Ther 1992; 51: 239–248.
- Guidance for Industry: Drug metabolism/drug interactions in the drug development process: studies in vitro. http://www.fda.gov/cder/guidance.

