В статье описываются результаты расчета ряда моделей метиламинов на основе теории функционала плотности и применение метода наименьшего градиента плотности для исследования нековалентных взаимодействий.
Ключевые слова: метиламин, этиламин, диметиламин, триметиламин, дескрипторы, МО, электрофильность, DFT, RDG.
Исследуемая группа составлена из аминопроизводных с формулой
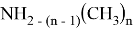
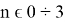
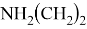
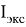
В частности, диметиламин и триметиламин являются предшественниками промышленно значимых соединений. Из-за несимметричного расположения азота в амине ряд соединений является хорошим акцептором электронов [4], следовательно была предложена возможность исследования связи «электронное строение — свойства» между квантово-химическими дескрипторами (нуклеофильность и электрофильность [5]) и электронной структурой молекулярных орбиталей (МО) в метиламинах.
Таблица 1
|
№ |
Молекула |
|
|
|
|
|
|
|
|
0 |
Аммиакат
|
10,15 |
10,59 |
17,85 |
28,45 |
0,46 |
2,16 |
0,13 |
|
1 |
Метиламин
|
9,66 |
9,88 |
15,41 |
25,29 |
0,30 |
3,31 |
0,18 |
|
2 |
Диметиламин
|
8,94 |
9,37 |
14,35 |
23,73 |
0,26 |
3,83 |
0,20 |
|
3 |
Триметиламин
|
8,5 |
8,93 |
13,58 |
22,52 |
0,24 |
4,15 |
0,21 |
|
4 |
Этиламин
|
9,50 |
9,44 |
15,01 |
24,76 |
0,28 |
3,56 |
0,19 |
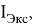
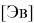
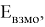
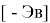
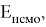
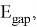
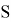
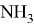
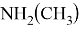
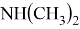
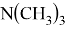
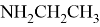
Для ряда
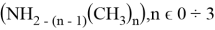
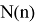
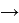
![a) Энергия ионизация орбитали N(n) и O(n) MO для формальдегидов (O= и n-Амины (H-N = , штриховые линии обозначают изменения I в открытых соединениях [6]; b) Энергия ионизации орбитали N(n) MO в метиламине](https://articles-static-cdn.moluch.org/articles/j/103703/images/103703.022.png)
Рис. 1. a) Энергия ионизация орбитали N(n) и O(n) MO для формальдегидов (O=
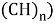
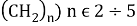
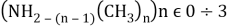
Рассчитанные значения внутреннего электронного распределения молекул, используются на практике для приблизительного определения химических характеристик. Квантово-химические дескрипторы отражают определенные свойства и тенденции в случае сложных соединений. В основе определения дескрипторов лежит величина энергетического разрыва между энергиями верхней занятой (
ВЗМО
(eng.
HOMO
)) и нижней свободной (
НСМО
(eng.
LUMO
)) молекулярных орбиталей (
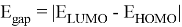
Абсолютная электроотрицательность молекул:
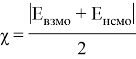
Также электрофильность
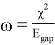
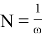
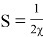
Отсюда было замечено, что индекс электрофильности метиламинов уменьшается при сцеплении каждого радикала метила (и, следовательно нуклеофильность увеличивается, что совпадает с их ролью как акцепторов в производстве сложных комплексов [4]).
Рассчитанные волновые функции МО (
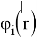
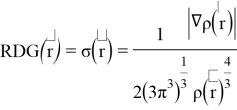
Где с
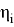
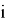
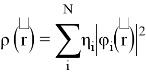
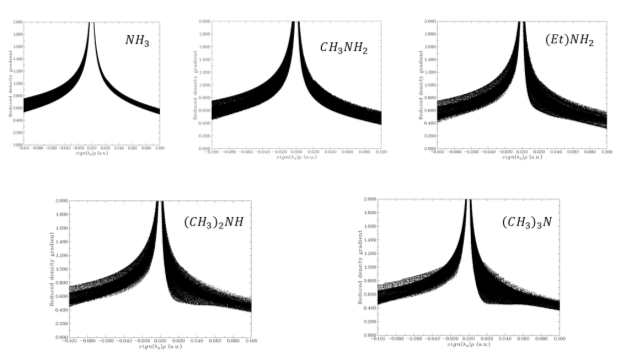
Рис. 2. RDG график для метиламинов (Вертикальная ось RDG, горизонтальная ось — электронная плотность, умноженная на знак второго собственного значения градиента
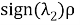
Было отмечено минимальное участие нековалентных взаимодействий. При значении
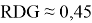
![NCI изоповерхности получены в программе VMD [9]](https://articles-static-cdn.moluch.org/articles/j/103703/images/103703.039.png)
Рис. 3. NCI изоповерхности получены в программе VMD [9]
Литература:
- The Role of Databases in Support of Computational Chemistry Calculations Feller, D., J. Comp. Chem. 1996, 17(13), 1571–1586.
- Barca, Giuseppe M. J. and Bertoni, Colleen and Carrington, Laura and Datta, Dipayan and De Silva, Nuwan and Deustua, J. Emiliano and Fedorov, Dmitri G. and Gour, Jeffrey R. and Gunina, Anastasia O. and Guidez, (…) Mark S. GAMESS & Recent developments in the general atomic and molecular electronic structure system // The Journal of Chemical Physics -2020, p154102, DOI:10.1063/5.0005188
- Вовна В.И, Вилесов Ф.И // Оптика и спектроскопия. 1974.Т.36, № 3. С 436–438.
- Ashford's Dictionary of Industrial Chemicals, 3rd edition, 2011, p 3284–3286.
- Electrophilicity Index, Robert G. Parr, La´szlo´ v. Szentpa´ly and Shubin Liu J. Am. Chem. Soc. 1999, vol 121, 9.
- В. И. Вовна. Электронная структура органических соединений по данным фотоэлектронной спектроскопии, Москова «НАУКА», 7, 115–114 (1991).
- Absolute electronegativity and hardness correlated with molecular orbital theory, Ralph G. P. // Proc. Natl. Acad. Sci. USA, V. 83, p 8440–8441.
- Tian Lu, Qinxue Chen. Mwfn: A Strict, Concise and Extensible Format for Electronic Wavefunction Storage and Exchange // ChemRxiv (2020) DOI: 10.26434/chemrxiv.11872524.
- VMD a molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting. https://www.ks.uiuc.edu/Research/vmd.

