В данной статье отражены классические морфологические проявления синдрома Альпорта на примере данных 18 нефробиопсий детей от 4 до 18 лет с данной патологией.
Ключевые слова: гломерулонефрит, нефробиопсия, синдром Альпорта.
СиндромАльпорта (СА)— этиологически гетерогенное наследственное заболевание моногенной природы. Причина заболевания лежит в мутации одного из генов: COL4A5, COL4A4, COL4A3. При классическом варианте СА мутация происходит в гене COL4A5, расположенном на длинном плече Х-хромосомы (Хq22.2), который кодирует 5-цепь коллагена IV типа (НГ).
Распространенность СА составляет 1:5000. Он лежит в основе развития 1 % всех случаев хронической почечной недостаточности (ХПН) в Европе. 2,3 % случаев трансплантации почки проводится больным с СА [1].
Классические изменения почечной паренхимы при синдроме Альпорта представляют сочетание морфологических черт хронического гломерулонефрита, пиелонефрита и интерстициального нефрита, но с неполной картиной каждой из патологий. Почки уменьшены в размерах, их структура мелкозернистая или палисадная. На срезе коркового вещества часто имеются желтые линейные прожилки. Первичные изменения наблюдаются в гломерулярной мембране. Наиболее ранним поражением являются выраженная пролиферация эпителия в клубочках, интерстициальный фиброз или локальное расширение с атрофией канальцев, появляющиеся приблизительно одновременно. В большинстве случаев наблюдаются заполненные липидами пенистые клетки, не связанные с эпителиальными клетками канальцев. Эти пенистые клетки могут заполнять интерстициальную ткань, особенно в нижних отделах коры, располагаясь в виде рядов или гнезд. В большинстве случаев в интерстициальной ткани находят плазматические клетки, лимфоциты и отложения кальция. Канальцевые изменения включают атрофию эпителия и расширение некоторых канальцев. Иногда наблюдается хронический интерстициальный нефрит, при котором изначально мало клубочков.
При анализе нефробиопсий 18 пациентов на базе патологоанатомического бюро 3-ей городской детской клинической больницы г. Минска в большинстве случаев (83 %) обнаружен диффузный или фокальный, глобальный или сегментарный мезангиопролиферативный гломерулонефрит. У 17 % обследованных выявленные изменения расценены как фокально-сегментарный гломерулосклероз. Тубулоинтерстициальные изменения включали обнаруженные во всех случаях пенистые клетки, одиночные и сформировавшиеся в виде кластеров в интерстиции и канальцах, 39 % случаях — очаговый межуточный склероз с атрофией канальцев, в 28 % случаев — очаговое межуточное воспаление разной степени выраженности. При иммуногистохимии в большинстве случаев (61 %) экспрессия иммуноглобулинов A, G, M и С3 С1q фракций комплемента отсутствовала, в 33 % случаев выявлена экспрессия иммуноглобулина M, в 11 % случаев — экспрессия иммуноглобулина M и С3 С1q фракций комплемента. У 8 пациентов, которым проведено иммуногистохимическое исследование с применением антител к α3 и α5 субъединицам коллагена IV типа, выявлена гетерогенность иммуногистохимического окрашивания — от полного отсутствия до сохранения обеих субъединиц.
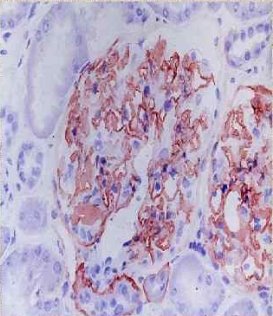
Рис. 1. Иммуногистохимическое исследование с применением антител к α3 (контроль)
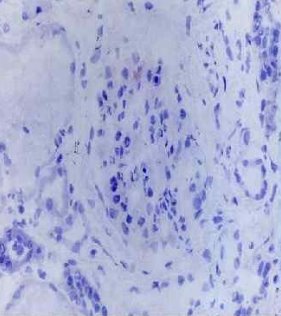
Рис. 2. Иммуногистохимическое исследование с применением антител к α3 (СА)
Таким образом, синдром Альпорта в большинстве случаев морфологически проявляется мезангиопролиферативным гломерулонефритом с наличием пенистых клеток и отсутствием экспрессии иммуноглобулинов A, G, M и С3 С1q фракций комплемента.
Литература:
- Atkin C. L., Gregory V. S., Border W. A. Alport syndrome. In: R. W. Schrier, C. M. Cottschalk (eds.) Diseases of the Kidney. 4th ed.Boston: Little 1989; 233.

