




Исследование комплексообразования мeди (II) С 2,6-дитиол-фенолом и гидрофобными аминами

Авторы: Кулиев Керим Аваз оглы, Вердизаде Наиля Аллахверди кызы, Исмаилова Шахназ Юсиф кызы
Рубрика: 5. Аналитическая химия
Опубликовано в
II международная научная конференция «Современная химия: Успехи и достижения» (Чита, апрель 2016)
Дата публикации: 07.04.2016
Статья просмотрена: 386 раз
Библиографическое описание:
Кулиев, К. А. Исследование комплексообразования мeди (II) С 2,6-дитиол-фенолом и гидрофобными аминами / К. А. Кулиев, Н. А. Вердизаде, Ш. Ю. Исмаилова. — Текст : непосредственный // Современная химия: Успехи и достижения : материалы II Междунар. науч. конф. (г. Чита, апрель 2016 г.). — Чита : Издательство Молодой ученый, 2016. — С. 16-23. — URL: https://moluch.ru/conf/chem/archive/162/9873/ (дата обращения: 25.04.2024).
Complex formation of copper with 2,6-dithiolphenol (DTF) and a hydrophobic amine (Am) was Investigated spectrophotometrically. Aniline (An), N-methylaniline (mAn), and N, N-dimethylaniline (dAn) were used as the hydrophobic amine. It has been found that ternary complexes formed in a weakly acidic medium (pHopt = 4.3–5.5). The ratio of the reactants in the mixed-ligand complexes corresponds to the Cu (II): DTP: Am = 1: 2: 4. The molar absorbance coefficients are equal to (2.92–3.35) • 104. Based on findings, procedure were developed for photometric determination of copper in different brands of steels and in oil and oil-products..
Keywords: copper, extraction-photometric method, 2,6-dithiollphenol, determination
Медь относится к числу тех металлов, которые обладают хромофорными свойствами, поэтому среди многочисленных фотометрических методов определения меди имеются как методы, основанные на использовании окрашенных реагентов с хромофорными группами, так и методы, в которых применяют бесцветные реагенты. Большинство методов обладают высокой избирательностью. Это дитизоновый, дитиокарбоматный, купроиновый и купризоновый методы или метод, с применением бис-(циклогексанон)оксалилдигазона; наибольшей чувствительностью обладает дитизоновый метод [1]. Реагенты, содержащие гидрокси — и карбокси -, или две гидрокси-группы в орто положении друг к другу, взаимодействуют с медью преимущественно в слабокислых и нейтральных средах с образованием окрашенных комплексных соединений [2].
Для экстракционно-спектрофотометрическое определения меди (II) предложен изонитрозопропиофенон [3]. Разработан чувствительная и селективная методика спектрофотометрического определения меди (II) в сплавах и в воде [4].
В неионно- мицеллярной среде фотометрическим методом определяли следовых количеств Cu (II) с 1- (2-пиридилазо) -2-нафтолом (ПАН) в присутствии нейтрального поверхностно-активного вещества, Тритон Х-100. [5].
Для спектрофотометрического определения меди в некоторых экологических и биологических объектах с помощью предложен салицилальдегид бензоил гидразон [6]. Разработаны экстракционно-фотометрическая методика определения меди с ализариновым желтым Р и триизобутил-фосфатом в пищевых продуктах [7].
Cогласно гипотезы аналогий, реакции с реагентами типа R-SH возможны для ионов элементов, образующих малорастворимые в воде сульфиды [8]. Разработаны методики определения элементов в виде разнолигандных комплексов (РЛК) с 2-гидрокси-5-галогентиофе-нолами и гидрофобными аминами [9,10, 11]. Представляло интерес изучение комплексообразования мeди (II)с 2,6-дитиолфенолом.
Представленная работа посвящена спектрофотометрическому исследованию взаимодействия меди с 2,6-дитиолфенолом в присутствии анилина, N-метиланилина, и N,N-диметиланилина.
Экспериментальная часть
Реагенты и растворы. Для приготовленияисходного раствора (1мг/мл) меди 3,9296г свежеперекристаллизованный CuSO45H2Oрастворили в воде, прибавляли 2 капли конц. серной кислоты и разбавляли в мерной колбе водою до 1л [12]. Растворы с концентрацией 0,1 мг/мл получали разбавлением исходного раствора. В работе использовали 0,01М раствор 2,6-дитиолфенола (ДТФ) и гидрофобных аминов (Ам). Анилин (Ан), N — метиланилин (мАн) и N,N-диметиланилин (дАн) использовали в свежеперегнанном виде. В качестве экстрагента применен очищенный хлороформ.
Ионную силу растворов, равную µ=0,1 поддерживали постоянной, введением рассчитанного количества KCl. Для создания необходимой кислотности растворов применяли 0,1М раствор HCI.
Аппаратура. Оптическую плотность органической фазы измеряли на КФК-2 и СФ-26. Значение рН водной фазы контролировали при помощи прибора И-120.2. со стеклянным электродом. ИК-спектры снимали на спектрофотометре UR-20. Все указанные приборы прошли госпроверку.
Методика. Вградуированные пробирки с притертыми пробками вводили 0,1–0,8 мл, с интервалом 0,1 мл исходного раствора меди, 2,2 мл 0,01М раствора ДТФ и 0,6–0,8 мл Ам. Необходимое значение рН устанавливали добавлением 2 мл 0,1М раствора NaOH. Объем органической фазы доводили до 5 мл хлороформом, а водной фазы — до 20 мл дистиллированной водой. Спустя 5 минут органический слой отделяли и измеряли его оптическую плотность при комнатной температуре на КФК-2 при 490 нм.
Результаты иих обсуждение
Представленная работа посвящена спектрофотометрическому исследованию взаимодействия меди (II) с 2,6-дитиоленолом. ДТФ с медью образует окрашенный комплекс, нерастворимый в неполярных органических растворителях. Заряд комплекса был установлен методом электромиграции ионов и методом электрофореза на бумаге. При изучении электро-миграции данного комплекса, в U-об-разной трубке наблюдалось движение окрашенных в оранжевый цвет ионов к положительному полюсу, на основании чего был сделан вывод о том, что окрашенный комплекс является анионом. При введении в систему гидрофобных аминов наблюдается экстракция анионного комплекса в органическую фазу в виде разнолигандного комплекса (РЛК). Из гидрофобных аминов использованы анилин, N-метиланилин, N,N-диметиланилин. На основании полученных данных разработаны новые избирательные и высокочувствительные методики фотометрического определения микроколичеств меди в сталях различных марок.
Выбор экстрагента. Для экстракции РЛК использованы неводные растворители: хлороформ, 1,2-дихлорэтан, четыреххлористый углерод, бензол, хлорбензол, толуол, ксилол, изобутанол, изопентанол и диэтиловый эфир. Экстрагируемость комплексов оценивали коэффициентом распределения и степенью экстракции. Наилучшими экстрагентами оказались хлороформ, дихлорэтан и четыреххлористый углерод. При однократной экстракции хлороформом извлекается 98,2–98,6 % меди в виде РЛК. Дальнейшие исследования проводили с хлороформом. Содержание меди в органической фазе определяли фотометрически — дитиолом после реэкстракции, а в водной — по разности. При экстракции комплексов меди с ДТФ молекулы хлороформа не входят в состав экстрагирующихся комплексов; сольватное число, характеризующееся значением тангенса угла наклона в данном случае равно нулю. При уменьшении диэлектрической проницаемости растворителя константы устойчивости комплексов возрастают. Понижение растворимости реагентов и комплексов в воде, как правило, сопровождается улучшением их растворимости в органических растворителях. Применение экстракционных методов намного повышает избирательность и чувствительность реакций.
Влияние рН водной фазы. Для образования и экстракции смешанных комплексов оптимальным является рН=4,9–6,1. При рН раствора ![]() 7 экстракция комплексов практически не наблюдается, что, видимо, связано с понижением степени протонизации аминов. С другой стороны увеличивается концентрация в водном растворе неэкстрагирующегося комплекса [Сu(ДТФ)2]4-, так как диссоциация ДТФ по второй сульфгидрильной группе (рК2=8,56) продолжает возрастать.
7 экстракция комплексов практически не наблюдается, что, видимо, связано с понижением степени протонизации аминов. С другой стороны увеличивается концентрация в водном растворе неэкстрагирующегося комплекса [Сu(ДТФ)2]4-, так как диссоциация ДТФ по второй сульфгидрильной группе (рК2=8,56) продолжает возрастать.
Зависимость оптической плотности от рН представлена на рис.1. Наличие одного максимума оптической плотности в указанных пределах рН подтверждает предположение об образовании одного комплексного соединения.

Рис.1. Зависимость оптической плотности РЛК меди (II) от рН водной фазы
1.Cu — ДТФ — Ан, 2.Cu — ДТФ — мАн, 3.Cu — ДТФ — дАн
![]() = 1,87510–5 М; СДТФ=0,8810–3 М;
= 1,87510–5 М; СДТФ=0,8810–3 М; ![]() = 1,2410–3 М, КФК-2, =540нм, l=0.5 cм.
= 1,2410–3 М, КФК-2, =540нм, l=0.5 cм.
Влияние концентрации лигандов и времени выдерживания. РЛК меди образуются в присутствии большого избытка комплексообразующих реагентов. Оптимальным условием образования и экстракции этих соединений является 0,8810–3 моль/л концентрация ДТФ и 1,2410–3 моль/л — Ам.
РЛК меди с ДТФ и Ам устойчивы в водных и органических растворителях и не разлагаются в течении трех суток, а после экстракции — больше месяца. Максимальная оптическая плотность достигается в течении 5 минут.
Спектры поглощения. Максимальный аналитический сигнал при комплексообразовании меди с ДТФ и Ам наблюдается при 530–536 нм (рис.2). ДТФ максимально поглощает при 270 нм. При комплексообразовании наблюдается батохромное смещение максимума светопоглощения на 260–266 нм. Контрастность реакций высока: исходные реагенты почти бесцветны, а комплексы-красно-бурого цвета. Молярные коэффициенты поглощения составляют (2,92 -3,35)104.
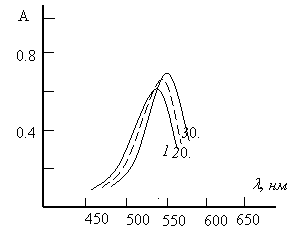
Рис.2. Спектры поглощения комплексов меди(II) с ДТФ и Ам
1. Cu -ДТФ-Ан, 2.Cu -ДТФ-мАн, 3.Cu -ДТФ-дАн
![]() = 1,875 10–5 М; СДТФ= 0,88 10–3 М;
= 1,875 10–5 М; СДТФ= 0,88 10–3 М; ![]() = 1,2410–3 М, СФ-26, l=1cм.
= 1,2410–3 М, СФ-26, l=1cм.
Состав и строение комплексов. Стехиометрию исследуемых комплексов устанавливали методами сдвига равновесия и относительного выхода[13]. Приведенные на рис.3 данные показывают, что соотношение компонентов в составе РЛК Cu(II): ДТФ: Ам. Методом Назаренко было установлена, что комплексообразующей формой меди является Cu2+[14,15]. При этом число протонов, вытесняемых им из одной молекулы ДТФ, оказалось равным 1.
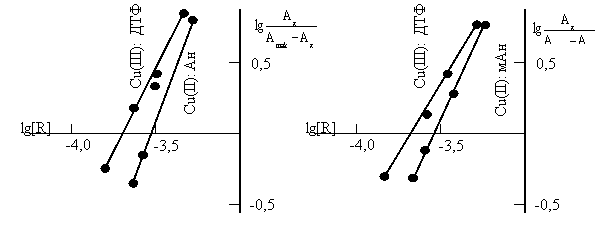
Рис.3. Определение состава комплекса Cu -ДТФ-Ам методом сдвига равновесия
![]() = 1,875 10–5 М; СФ-26, l=1cм.
= 1,875 10–5 М; СФ-26, l=1cм.
В ИК-спектрах комплекса в области 950–960 см-1 появляется интенсивная полоса поглощения, которая отсутствует в спектрах реагента. Эта полоса обусловлена валентным колебанием связи метал-лиганд. Исчезновение ярко выраженной полосы при 2580см-1, наблюдаемая в спектре ДТФ и появление в спектрах комплекса двух полос поглощения, одна из которых смещена в сторону меньших частот, говорит о том, что один из –SH групп участвует в образовании комплекса. Исчезновение полосы поглощения в области 3200–3600см-1 с максимумом при 3460см-1 показывает, что гидроксильная группа принимает участие в образовании связи с металлом. Обнаружение полосы поглощения при 2380 см-1 указывает на наличие протонированного анилина, а полосы поглощения при 1370 см-1 — на наличие координированного анилина [16,17].
Медь (II) с dsp2гибридными орбиталями образует плоские квадратные комплексы. Однако, плоские квадратные комплексыCu(II) с одним неспаренным электроном способны присоединить дополнительные лиганды, образуя октаэдрические комплексы [2].
Произведенные расчеты показали, что РЛК в органической фазе не полимеризуются и находятся в мономерной форме (=1,05–1,08) [18].
Механизм образования РЛК можно представить следующим образом. Ионы меди при взаимодействии с двумя молекулами ДТФ и Ам образуют двухзарядный анионный комплекс, который экстрагируется с двумя молекулами протонированного Ам. Состав экстрагируемых смешанных комплексов можно представить формулой ![]() .
.
Можно предположить, что при комплексообразовании происходят процессы:
![]() (1)
(1)
![]()
![]() (2)
(2)
Константа равновесия реакции равна![]()
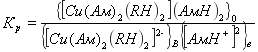 (3)
(3)
Поскольку коэффициент распределение (D) равен
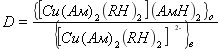 (4)
(4)
То
![]() (5)
(5)
Прологарифмировав последнее выражение, получим
![]() (6)
(6)
Величины ![]() , вычисленные по формуле(6) для комплексов [Cu(ДТФ)2(Ан)2](АнН)2 [Cu(ДТФ)2(мАн)2](мАнH)2 и [Cu(ДТФ)2(дАн)2](дАнH)2 равны 6,55±0,27; 6,62±0,14 и 6,79±0,34 соответс-твенно.
, вычисленные по формуле(6) для комплексов [Cu(ДТФ)2(Ан)2](АнН)2 [Cu(ДТФ)2(мАн)2](мАнH)2 и [Cu(ДТФ)2(дАн)2](дАнH)2 равны 6,55±0,27; 6,62±0,14 и 6,79±0,34 соответс-твенно.
В табл. 1. приведены основные спектрофотометрические характеристики РЛК меди.
Таблица 1
Характеристика РЛК меди сДТФ иАм.
|
Соединение |
рН |
,нм |
,нм |
·10–4 л·мол-1·см-1· |
Рабочий диапазон, мкг/мл |
|
|
Образования и экстракции |
Оптимальная |
|||||
|
[Cu(ДТФ)2(Ан)2](АнН)2 |
1.5–5.7 |
4.3–5.2 |
530 |
260 |
2.92 |
0.05–3.2 |
|
[Cu(ДТФ)2(мАн)2](мАнH)2 |
1.8–5.9 |
4.4–5.3 |
534 |
264 |
3.21 |
0.05–3.2 |
|
[Cu(ДТФ)2(дАн)2](дАнH)2 |
2.0–6.4 |
4.5–5.5 |
536 |
266 |
3.35 |
0.05–3.4 |
В интервале концентраций 0,05–3.4 мкг в 1мл экстракта ионных ассоциатов соблюдается линейная зависимость между оптической плотностью и концентрацией раствора.
Влияниепосторонних ионов. Для оценки применимости экстрактов РЛК для разделения и определения меди изучено мешающее влияние посторонних ионов. Избирательность спектрофотометрического определения меди в виде изученных комплексов представлена в табл.2. Установлено, что большие количества щелочных, щелочноземельных элементов, РЗЭ, F-, CI-, ![]() не мешают определению меди. Олределению мешают цитраты и тартраты, J-,CN-,
не мешают определению меди. Олределению мешают цитраты и тартраты, J-,CN-, ![]() , тиомочевина. Мешающее влияние Fе (III) устраняли щавеловой кислотой; Ti(IV)– фторидом натрия или тайроном; Hg(II)-сульфит ионом; Nb(V) и Ta(V) –щавелевой кислотой, а Mo(VI) и W(VI)- фторидом натрия и щавелевой кислотой. При использовании 1 %-го раствора аскорбиновой кислоты определению не мешают Mn(VII), V(IV), Nb(V), Cr(VI), Mo(VI) и Fe(III). При использовании 0,01М раствора щавеловой кислоты определению не мешают V(IV), Nb(V), Ta(V), Cr(III), Mo(VI), W(VI) и Fe(III). Кроме того Nb(V), Ta(V) и Ti(IV) с ДТБФ и Ам образуют комплексы в более кислой среде.
, тиомочевина. Мешающее влияние Fе (III) устраняли щавеловой кислотой; Ti(IV)– фторидом натрия или тайроном; Hg(II)-сульфит ионом; Nb(V) и Ta(V) –щавелевой кислотой, а Mo(VI) и W(VI)- фторидом натрия и щавелевой кислотой. При использовании 1 %-го раствора аскорбиновой кислоты определению не мешают Mn(VII), V(IV), Nb(V), Cr(VI), Mo(VI) и Fe(III). При использовании 0,01М раствора щавеловой кислоты определению не мешают V(IV), Nb(V), Ta(V), Cr(III), Mo(VI), W(VI) и Fe(III). Кроме того Nb(V), Ta(V) и Ti(IV) с ДТБФ и Ам образуют комплексы в более кислой среде.
Таблица 2
Влияние посторонних ионов на определение меди сДТФ иАм. (взято 30мкг Cu(II))
|
Ион |
Мольный избыток иона |
Маскирующий реагент |
Найдено,мкг |
RSD (%) |
|
Co(II) |
50 |
|
30,0 |
2 |
|
Ni(II) |
50 |
|
29,8 |
2 |
|
Fe(II) |
200 |
|
29,8 |
3 |
|
Cd(II) |
200 |
|
29,6 |
4 |
|
Al(III) |
180 |
30,0 |
2 |
|
|
Fe(III) |
60 |
Щавелевая кислота |
30,2 |
4 |
|
Zr(IV) |
50 |
29,8 |
3 |
|
|
W(VI) |
25 |
Щавелевая кислота |
29,6 |
5 |
|
Hg(II) |
30 |
30,2 |
5 |
|
|
Ti(IV) |
30 |
тайрон |
29,6 |
3 |
|
V(IV) |
20 |
тайрон |
29,6 |
3 |
|
Mo(VI) |
10 |
Фторид натрия |
30,4 |
4 |
|
Cr(III) |
120 |
29,8 |
4 |
|
|
Nb(V) |
50 |
Фторид натрия |
30,1 |
5 |
|
Ta(V) |
50 |
Фторид натрия |
30,1 |
5 |
|
|
50 |
|
29,2 |
4 |
Относительное стандартное отклонение на основе трех определений
На основании уравнений градуировочных графиков рассчитывали предел фотометрического обнаружения и предел количественного определения меди в виде ионных ассоциатов [19].
В таблице 3 приведены калибровочные характеристики тройных комплексов Сu-ДТФ-Ам.
Таблица 3
Аналитические характеристики для тройных комплексов Сu-ДТБФ-Ам
|
Параметр |
Сu-ДТФ-Ан |
Сu-ДТФ-мАн |
Сu-ДТФ-дАн |
|
Уравнение градуировочных графиков |
у = 0,035 + 0,425x |
у = 0,032 + 0,466 x |
у = 0,039 + 0,489x |
|
Коэффициент корреляции |
0.9993 |
0.9995 |
0.9995 |
|
Линейный диапазон градуировочных графиков, мкг/мл |
0.05–3.2 |
0.05–3.2 |
0.05–3.4 |
|
Предел обнаружения нг/ см3 |
10,50 |
9,70 |
8,60 |
|
Предел количественного определения, нг/ см3 |
34,65 |
32,31 |
28,38 |
|
Чувствительность, нг/ см2 |
2,19 |
1,99 |
1,91 |
В табл. 4 приведены данные, позволяющие сравнить аналитические характеристики методик определения меди с некоторыми уже известными [1] методиками.
Таблица 4
Сравнительные характеристики методик определения меди
|
Реагент |
pH(растворитель) |
, нм |
10–4 |
|
|
Известные методики |
||||
|
Диэтилдитиокарбаминат |
4–11(четыреххлористый углерод) |
436 |
1,4 |
|
|
Купроин |
4–7(изоамиловый спирт) |
546 |
0,64 |
|
|
Неокупроин |
3–10(изоамиловый спирт) |
454 |
0,79 |
|
|
Предлагаемые методики |
||||
|
Дтф+мАн |
4.4–5.3 (хлороформ) |
534 |
3,21 |
|
|
ДТФ+дАн |
4.5–5.5 (хлороформ) |
536 |
3,35 |
|
Определение меди в сталях. Навеску стали (около1г) растворяли в 10 мл смеси концентрированных кислот HCl и HNO3 в соотношении 3:1. После полного растворения добавляли 5 мл H2SO4(1:1) и полученный раствор выпаривали до прекращения выделения паров SO3. Прибавляли 30 мл воды, 2–3 мл 30 % раствора H2O2 и нагревали до кипения. Нерастворимый осадокотфильтровывали через сухой бумажный фильтр, осадок промывали дважды дистиллированной водой. Фильтрат и промывные воды собирали в мерную колбу емкостью 100 мл и после охлаждения разбавляли дистиллированной водой до метки. Отбирали аликвотную часть полученного раствора, переносили в делительную воронку, создали рН 5 и 2,2 мл 0,01 М дтф. После тщательного перемешивания прибавляли 2,8 мл 0,01 М мАн. Объем органической фазы доводили до 5 мл хлороформом, а общий объем — до 25 мл дистиллированной водой. Смесь встряхивали 5 мин. После расслаивания фаз светопоглощение экстрактов измеряли на КФК-2 при 540 нм в кювете с толщиной 0,5 см. Содержание меди находили по градуировочному графику.
Результаты экспериментов представлены в табл. 5. Как видно из таблицы, результаты определения меди в стали свидетельствуют о достаточной надежности предлагаемых методик.
Таблица 5
Правильность ивоспроизводимость определений меди встали (с16б, Cu-0,219%). n=6, Р=0,95
|
Метод |
|
Сходимость,% |
SD |
RSD,% |
|
|
Диэтилдитио-карбаминат |
0,214 |
98,5 |
0,0120 |
5,6 |
0,214±0,012 |
|
Дтф+мАн |
0,216 |
99,0 |
0,0110 |
5,1 |
0,216±0,011 |
|
ДТФ+дАн |
0,220 |
101,5 |
0,0094 |
4,3 |
0,220±0,010 |
Определение меди внефти. Для определения содержания меди в нефти в фарфоровую чашку с диаметром 90 мм брали с точностью 0,001г навеску 40г испытуемого топлива и сжигали в соответствии ГОСТ 1461–75. Затем фарфоровую чашку с золой помещали в муфель при температуре 550±20оС и выдерживали при этой температуре 1 час. После охлаждения в чашку добавляли 5 мл HCl (1:1), кипятили ее содержимое до объема 2мл и добавляли к нему 0,5г безводного Na2CO3; затем чашку помещали на 2–3 минуты в муфель, нагретый до 800оС. После охлаждения остаток в чашке растворяли в 5 мл дистиллированной воды, дважды фильтровали через один и тот же фильтр в мерную колбу с вместимостью 50 мл, промывали осадок 2–3 раза дистиллированной водой, промывные воды присоединяли к фильтрату и нагревали до объема около 25 мл, переносили в колбу с емкостью 25 мл, подкисляли прибавлением 1–2 мл конц. HCl и объем раствора довели до метки дистиллированной водой. Отбирали аликвотные части и добавляли к ним одинаковые объемы сульфата меди (30 мкг Cu(II)). Содержание меди определяли разработанными экстракционно-спектрофотометрическими методами по прописи, описанной при построении градуировочных графиков. Разработанные методики определения меди контролировали фотометрически — диэтилдитиокарбаминатом.
Правильность и воспроизводимость определения меди в нефти показаны в табл. 6.
Таблица 6
Правильность ивоспроизводимость определений меди внефти (n=6, Р=0,95)
|
Метод |
|
Сходимость,% |
S |
Sr |
|
|
Калмас СК № 416 |
|||||
|
Диэтилдитио-карбаминат |
1,12·10–2 |
98,5 |
0,000381 |
0,034 |
(1.12±0.04)·10–2 |
|
Дтф+Ан |
1,14·10–2 |
99,0 |
0,000296 |
0,026 |
(1.14±0.03)·10–2 |
|
ДТФ+дАн |
1,15·10–2 |
101,5 |
0,000368 |
0,032 |
(1.15±0.04)·10–2 |
|
Грязевая сопка СК № 416 |
|||||
|
Диэтилдитио-карбаминат |
1,16·10–2 |
102 |
0,00045 |
0,039 |
(1.16±0.05)·10–2 |
|
Дтф+Ан |
1,18·10–2 |
101 |
0,00033 |
0,028 |
(1.18±0.03)·10–2 |
|
ДТФ+мАн |
1,19·10–2 |
99 |
0,00045 |
0,024 |
(1.19±0.05)·10–2 |
|
НГД СК № 318 |
|||||
|
Диэтилдитио-карбаминат |
1,46·10–2 |
104 |
0.00054 |
0,037 |
(1.46±0.06)·10–2 |
|
ДТФ+Ан |
1,45·10–2 |
102 |
0.00056 |
0,038 |
(1.45±0.07)·10–2 |
|
Дтф+мАн |
1,49·10–2 |
98 |
0.00054 |
0,043 |
(1.49±0.06)·10–2 |
|
СК № 129 |
|||||
|
Диэтилдитио-карбаминат |
1,24·10–2 |
98 |
0.00059 |
0.048 |
(1.24±0.06)·10–2 |
|
ДТФ+мАн |
1,28·10–2 |
98 |
0.00049 |
0.039 |
(1.28±0.05)·10–2 |
|
Дтф+дАн |
1,29·10–2 |
103 |
0.00057 |
0.044 |
(1.29±0.06)·10–2 |
Литература:
- Марченко З., Бальцежак М. К. Методы спектрофотометрии в УФ и видимой областях в неорганическом анализе. М.:Бином. Лаборатория знаний. 2007. 711с.
- Умланд Ф., Янсен А., Тириг Д., Вюнш Г. Комплексные соединения в аналитической химии. М.: Мир, 1975. С. 286.
- Archana R Kocharekar and N V Takkar //Journal of Sciens & Industrial Research. Vol. 63. 2004. p. 283
- D. Rekha, K. Suvardhan, K. Suresh Kumar, P. Reddyprasad, B. Jayaraj and P. Chiranjeev // J. Serb. Chem. Soc. (2007) 72 (3) p 299–310
- Agnihotri NK, Singh VK, Singh HB // Talanta. 1997; 45 (2), p 331–341.
- M. Jamaluddin Ahmed and Tasnima Zannat. // Pak. J. Anal. Environ. Chem. (2012), Vol. 13, No. 1. p 22–35
- Рустамов Н. Х., Рустамова У. Н. // Молодой ученый. 2012. № 8. С. 47–50.
- Кузнецов В. В. Применение органических аналитических реагентов в анализе неорганических веществ. М.: МХТИ.1972. 145 с.
- Вердизаде Н. А., Амрахов Т. И., Кулиев К. А., Залов А. З. //Журн. аналит. химии. 1997. Т.52. № 10. С.1042.
- Вердизаде Н. А., Залов А. З., Аллахвердиев М. А., Ибрагимов Г. И. //Тез. докл. межд. Конференции «Экстракция органических соединений». Воронеж, 2010. С.171.
- Вердизаде Н. А., Залов, Кулиев К. А., Абаскулиева У. Б., Ибрагимов Г. И. // Всероссийская конференция «Химический анализ», Тезисы докладов. М.: ИОНХ. 2008.С.97.
- Коростелев П. Т. Приготовление растворов для химико-аналитических работ. М.: Изд-во АН СССР, 1964. 401с.
- Булатов М. И., Калинкин И. П. Практическое руководство по фотоколориметрическим и спектрофотометрическим методам анализа. Л.: Химия, 1986. 432c.
- Назаренко В. А., Бирюк Е. А. // Журн. аналит. химии.1967.Т.12. № 2. С. 463.
- Назаренко В. А. // Тр. комис. по аналит. химии АН СССР. М.: Наука, 1969. Т. 17. С.22.
- Накaмато К. ИК-спектры и спектры КР неорганических и координационных соединений. М.: Мир, 1991. 536c.
- Беллами Л. Инфракрасные спектры сложных молекул. М.:1963. 592с.
- Ахмедли М. К., Клыгин А. Е., Иванова Л. И., Баширов Э. А. // Журн. неорган. химии. 1974. Т. 19. № 8. С. 2007.
- Дорохова Е. Н., Прохорова Г. В. Аналитическая химия (физико-химические методы анализа). М.: Высшая школа, 1991. С. 250.
Похожие статьи
Установление состава комплекса по методу изомолярных серий...
III, медь, дистиллированная вода, комплекс, оптическая плотность, органическая фаза, фторид натрия, водная фаза, RSD, щавелевая кислота.
Экстракционно-фотометрическое определение меди...
III, медь, дистиллированная вода, комплекс, оптическая плотность, органическая фаза, фторид натрия, водная фаза, RSD, щавелевая кислота. Ключевые слова. определение, экстракционно-фотометрический метод, 2, медь...
Использование геохимических барьеров для очистки техногенных...
III, медь, дистиллированная вода, комплекс, оптическая плотность, органическая фаза, фторид натрия, водная фаза, RSD, щавелевая кислота. Исследование качественных реакций на катионы на внеурочных...
Исследование влияния добавок некоторых спиртов на...
III, медь, дистиллированная вода, комплекс, оптическая плотность, органическая фаза, фторид...
Определение электропроводности неводных и смешанных сред...
Однако такая особенность флотоэкстракции, как возможность многократной концентрации ионов металлов в небольших объемах органического растворителя вне
Извлечение короткоцепочечных жирных кислот из водных растворов метил-трет-бутиловым эфиром.
Синтез органических производных меди (II)
Органические производные двухвалентной меди — это, прежде всего, соли карбоновых кислот (карбоксилаты), а также различные комплексы
Осадок отфильтровывали на воронке с бумажным фильтром, промывали 20 мл дистиллированной воды и сушили на воздухе.
Флотоэкстракция ионов никеля из водных растворов
Основные термины (генерируются автоматически): органическая фаза, извлечение никеля, водная фаза, размер пузырьков, положительное влияние, органический растворитель, вод, расход
водная фаза, коэффициент распределения, кислота, метил-трет-бутиловый эфир...
О влиянии выбросов алюминиевого завода на содержание...
Поэтому в состав буфера для создания общей ионной силы добавлен цитрат натрия (Трилон Б), который вытесняет фторид-ионы из комплексов с металлами.
CF — концентрация фторид-ионов в водной вытяжке, моль/л
Кондуктометрическое определение железа | Статья в журнале...
III, универсальный буфер, титрование, щавелевая кислота, металл, калий, амперометрическое титрование, М раствора, форма кривых титрования
Как показал рентгенографический анализ, в полученных слитках имеется в основном одна фаза: линии на рентгенограммах сдвинуты...
Похожие статьи
Установление состава комплекса по методу изомолярных серий...
III, медь, дистиллированная вода, комплекс, оптическая плотность, органическая фаза, фторид натрия, водная фаза, RSD, щавелевая кислота.
Экстракционно-фотометрическое определение меди...
III, медь, дистиллированная вода, комплекс, оптическая плотность, органическая фаза, фторид натрия, водная фаза, RSD, щавелевая кислота. Ключевые слова. определение, экстракционно-фотометрический метод, 2, медь...
Использование геохимических барьеров для очистки техногенных...
III, медь, дистиллированная вода, комплекс, оптическая плотность, органическая фаза, фторид натрия, водная фаза, RSD, щавелевая кислота. Исследование качественных реакций на катионы на внеурочных...
Исследование влияния добавок некоторых спиртов на...
III, медь, дистиллированная вода, комплекс, оптическая плотность, органическая фаза, фторид...
Определение электропроводности неводных и смешанных сред...
Однако такая особенность флотоэкстракции, как возможность многократной концентрации ионов металлов в небольших объемах органического растворителя вне
Извлечение короткоцепочечных жирных кислот из водных растворов метил-трет-бутиловым эфиром.
Синтез органических производных меди (II)
Органические производные двухвалентной меди — это, прежде всего, соли карбоновых кислот (карбоксилаты), а также различные комплексы
Осадок отфильтровывали на воронке с бумажным фильтром, промывали 20 мл дистиллированной воды и сушили на воздухе.
Флотоэкстракция ионов никеля из водных растворов
Основные термины (генерируются автоматически): органическая фаза, извлечение никеля, водная фаза, размер пузырьков, положительное влияние, органический растворитель, вод, расход
водная фаза, коэффициент распределения, кислота, метил-трет-бутиловый эфир...
О влиянии выбросов алюминиевого завода на содержание...
Поэтому в состав буфера для создания общей ионной силы добавлен цитрат натрия (Трилон Б), который вытесняет фторид-ионы из комплексов с металлами.
CF — концентрация фторид-ионов в водной вытяжке, моль/л
Кондуктометрическое определение железа | Статья в журнале...
III, универсальный буфер, титрование, щавелевая кислота, металл, калий, амперометрическое титрование, М раствора, форма кривых титрования
Как показал рентгенографический анализ, в полученных слитках имеется в основном одна фаза: линии на рентгенограммах сдвинуты...




