





Химия органических пероксидов насчитывает более ста лет своей истории. Ключевыми реагентами в синтезе пероксидов являются кетоны и альдегиды вследствие своей доступности и легкости протекания реакции между углеродным атомом карбонильной группы и высоконуклеофильным атомом кислорода гидропероксидной группы [1–4]. На протяжении последних трех десятилетий интенсивное развитие химии органических пероксидов в значительной степени связано с обнаружением у них высокой противомалярийной и антигельминтной активности [5–13]. Природный пероксид Артемизинин и его полусинтетические производные (Артеметер, Артеэтер, Артесунат) на протяжении двух десятилетий применяются для лечения малярии [14]. Органические пероксиды на основе кетонов производятся в крупнотоннажном масштабе и применяются в качестве инициаторов радикальной полимеризации и сшивки [15–19]. Потребность в недорогих и высокоэффективных, технологически доступных биологически активных соединениях и инициаторах полимеризации послужила мощным импульсом в развитии методов синтеза пероксидов различных классов из кетонов, их производных, пероксида водорода и гидропероксидов [20–29].
На сегодняшний день пероксидирование монокетонов и их производных изучено очень детально. Пероксидирование дикетонов изучено в меньшей степени, вероятно это связано с тем, что с увеличением числа карбонильных групп резко возрастает количество продуктов реакции. По этой причине селективный синтез пероксидов на основе дикетонов представляется трудно выполнимой задачей.
Ранее нами было обнаружено, что селективный синтез пероксидов, а именно мостиковых тетраоксанов, можно осуществлять на основе реакции β-дикетонов с H2O2, катализируемой сильными кислотами (H2SO4, HClO4, HBF4, и BF3·Et2O) и гетерополикислотами (фосфорномолибденовая, фосфорновольфрамовая) в гомогенных условиях [30–31]. Использование гетерополикислот в качестве катализаторов позволяет синтезировать тетраоксаны из β-дикетонов как незамещенных, так и замещенных по α-положению, а также получать тетраоксаны с алкильными, арильными и адамантильным заместителями в боковой цепи.
В настоящей работе метод синтеза мостиковых тетраоксанов был упрощен за счет проведения реакции в гетерогенных условиях. Использование в качестве катализатора силикагеля с нанесенной фосфорномолибденовой кислотой (ФМК) позволяет селективно и с хорошим выходом получать целевой пероксид, а также значительно упростить процедуру его выделения.
Оптимизацию условий получения тетраоксанов проводили на примере превращения 3-бутилацетилацетона 1 в тетраоксабицикло [2.2.1]гептан 2 с использованием трехкратного мольного избытка эфирного раствора H2O2 (М = 1,10 моль/л). В качестве катализатора использовали силикагель с нанесенной фосфорномолибденовой кислотой, массовое содержание которой составляло 10, 20 и 30 % соответственно (ФМК·SiO2). Мольное соотношение ФМК: дикетон 1 составляло 0,1:1. Исследовали влияние количества катализатора, растворителя и времени проведения реакции на выход тетраоксана 2 (таблица 1).
Таблица 1
Синтез тетраоксабицикло [2.2.1]гептана 2 из 3-бутилацетилацетона 1.
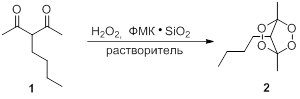
|
Опыт |
Тип катализатора ФМК·SiO2 (масс. % ФМК) |
Растворитель |
Время, ч |
Выход 2, % по ЯМР (на выделенный продукт) |
|
1 |
10 |
Этилацетат |
12 |
54 |
|
2 |
10 |
Этилацетат |
24 |
34 |
|
3 |
10 |
Диэтиловый эфир |
12 |
55 |
|
4 |
10 |
Диэтиловый эфир |
24 |
57 |
|
5 |
10 |
Толуол |
12 |
64 |
|
6 |
10 |
Толуол |
24 |
76 (66) |
|
7 |
10 |
ТГФ |
12 |
63 (52) |
|
8 |
10 |
ТГФ |
24 |
62 |
|
9 |
20 |
Этилацетат |
12 |
66 |
|
10 |
20 |
Этилацетат |
24 |
80 |
|
11 |
20 |
Диэтиловый эфир |
12 |
42 |
|
12 |
20 |
Диэтиловый эфир |
24 |
59 |
|
13 |
20 |
Толуол |
12 |
73 |
|
14 |
20 |
Толуол |
24 |
88 |
|
15 |
20 |
ТГФ |
12 |
47 |
|
16 |
20 |
ТГФ |
24 |
63 |
|
17 |
30 |
Этилацетат |
12 |
93 (84) |
|
18 |
30 |
Этилацетат |
24 |
86 (75) |
|
19 |
30 |
Диэтиловый эфир |
12 |
92 (80) |
|
20 |
30 |
Диэтиловый эфир |
24 |
60 |
|
21 |
30 |
Толуол |
12 |
98 (85) |
|
22 |
30 |
Толуол |
24 |
85 (70) |
|
23 |
30 |
ТГФ |
12 |
90 (79) |
|
24 |
30 |
ТГФ |
24 |
77 |
С использованием ФМК·SiO2 с 10 масс. % ФМК (опыты 1–8) наибольший выход (66 %) был достигнут за 24 часа с использованием в качестве растворителя толуола. При использовании ФМК·SiO2 с 20 масс. % ФМК (опыты 9–16) достигается выход в 88 % по ЯМР (опыт 14). Применение ФМК·SiO2 с 30 масс. % ФМК позволяет синтезировать тетраоксан 2 с высоким выходом и наилучший результат (98 % по ЯМР и 85 % на выделенный продукт) был получен в условиях опыта 21.
По результатам оптимизации можно заключить, что ключевыми параметрами, определяющими хороший выход тетраоксана 2,являются ФМК·SiO2 с 30 масс. % ФМК, мольное соотношение ФМК: дикетон 1 равное 0,1:1, трехкратный мольный избыток H2O2 ииспользование в качестве растворителя толуола.
Методика получения силикагеля с нанесенной фосфорномолибденовой кислотой:
В 15 мл. этанола растворяли 0,55; 1,25 или 2,14 г. (0,235; 0,534 или 0,914 ммоль) ФМК (фосфорномолибденовая кислота, 78 %). При перемешивании к полученному раствору добавляли 5 г силикагеля (SiO2) 60 / 200 (количество ФМК в ФМК·SiO2 составляет 10, 20 или 30 масс. % соответственно). Образовавшуюся суспензию подвергали интенсивному перемешиванию в течение 10 мин. для достижения количественной адсорбции молекул фосфорномолибденовой кислоты на частицах силикагеля. Во избежание образования конгломератов частиц, суспензию подвергали обработке ультразвуком в течение 5 мин. Затем растворитель упаривали в вакууме водоструйного насоса. Так как испарение растворителя может приводить к слипанию частиц, упаривание проводили в течение 5 мин. строго при давлении 6 мм.рт.ст. Контроль давления осуществляли при помощи вакуумметра. После упаривания растворителя полученную суспензию повторно обрабатывали ультразвуком в течение 5 мин, затем подвергали сушке в вакууме водоструйного насоса. Данную последовательность операций повторяли до прекращения изменения массы катализатора. Для гарантированного разрушения конгломератов полученный катализатор подвергали финальной обработке ультразвуком в течение 30 мин.
Методика синтеза тетраоксана 2:
К раствору 3-бутилацетилацетона 1 (0,3 г, 1,921 ммоль) в толуоле (5 мл) при перемешивании прибавляли эфирный раствор H2O2 (5,238 мл, 5,762 ммоль, 3 моль Н2О2 / 1 моль 3-бутилацетилацетона) и силикагель с нанесенной фосфорномолибденовой кислотой (ФМК·SiO2, 30 масс. % ФМК) (1,498 г, 0,1 моль ФМК / 1 моль 3-бутилацетилацетона). Перемешивали при 20–25°С в течение 12 часов. Реакционную смесь отфильтровывали, осадок промывали диэтиловым эфиром (2×30 мл). Органический слой промывали 5 % раствором NaHCO3 (10 мл) и затем водой (2×10 мл). Сушили над Na2SO4, фильтровали, удаляли растворитель в вакууме водоструйного насоса. Продукт тетраоксабицикло [2.2.1]гептан 2 выделяли хроматографией на SiO2, с использованием элюента петролейный эфир — этилацетат с увеличением доли последнего от 0 до 30 объемных процентов.
Аналитические характеристики тетраоксана 2:
Масло. Rf = 0.61 (ТСХ, гексан — этилацетат, 5:1).
Спектр ЯМР 1Н (300.13 МГц, δ, м.д): 0.92 (т, 3H, CH3, J = 6.6 Гц), 1.30–1.61 (м, 12H, CH2, CCH3), 2.59 (т, 1H, CH, J = 5.9 Гц).
Спектр ЯМР 13С (50.32 МГц, CDCl3, δ, м.д.): 9.75 (CH3CO), 13.75 (CH3CH2), 22.77 (CH3CH2 CH2), 23.51 (CH2CH), 29.69 (CH2CH2 CH2), 59.01 (CH), 110.79 (OCO).
Вычислено C9H16O4: C, 57.43; H, 8.57. Найдено: C, 57.30; H, 8.67.
Литература:
1. A. Baeyer, V. Villiger, Chem. Ber. 1900, 33, 858–864.
2. R. Criegee, W. Schnorrenberg, J. Becke, Justus Liebigs Ann. Chem. 1949, 565, 7–21.
3. V. L. Antonovskii, E. V. Fedorova, N. E. Shtivel’, Yu. D. Emelin, J. Org. Chem. USSR (English Translation) 1991, 27, 704–706, ЖурналОрганическойХимии 1991, 27, 820–823;
4. M. A. Avery, F. Gao, W. K. M. Chong, T. F. Hendrickson, W. D. Inman, P. Crews, Tetrahedron 1994, 50, 957–972.
5. C. W. Jefford, Curr. Top. Med. Chem. 2012, 12, 373–399.
6. R. D. Slack, A. M. Jacobine, G. H. Posner, MedChemComm. 2012, 3, 281–297.
7. I. Fernández, A. Robert, Org. Biomol. Chem. 2011, 9, 4098–4107.
8. N. Yadav, C. Sharma, S. K. Awasthi, RSC Adv. 2014, 4, 5469–5498.
9. A. O. Terent'ev, D. A. Borisov, I. A. Yaremenko, Chem. Heterocycl. Compd. 2012, 48, 55–58.
10. H. D. Hao, S. Wittlin, Y. Wu, Chem. Eur. J. 2013, 19, 7605–7619.
11. J. Boissier, J. Portela, V. Pradines, F. Coslédan, A. Robert, B. Meunier, C. R. Chim. 2012, 15, 75–78.
12. J. Keiser, K. Ingram, M. Vargas, J. Chollet, X. Wang, Y. Dong, J. L. Vennerstrom, Antimicrob. Agents Chemother. 2012, 56, 1090–1092.
13. K. Ingram, I. A. Yaremenko, I. B. Krylov, L. Hofer, A. O. Terent’ev, J. Keiser, J. Med. Chem. 2012, 55, 8700–8711.
14. World Health Organization, Guidelines for the treatment of malaria, WHO Press, 2010; b) World Health Organization, Management of severe malaria, WHO Press, 2013.
15. E. T. Denisov, T. G. Denisova, T. S. Pokidova, in Handbook of free radical initiators, John Wiley and Sons, 2003.
16. P. Nesvadba, in Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley and Sons, 2012;
17. H. F. Mark, in Encyclopedia of Polymer Science and Technology, Concise, 3rd Edition, John Wiley and Sons, 2007, pp-573–576.
18. P. Ray, Polymer Cross-Linkling. Encyclopedia Of Polymer Science and Technology, John Wiley and Sons, 2011.
19. H. W. Engels, H. J. Weidenhaupt, M. Pieroth, W. Hofmann, K. H. Menting, T. Mergenhagen, R. Schmoll, and S. Uhrlandt, Rubber, 9. Chemicals and Additives. Ullmann's Encyclopedia of Industrial Chemistry, John Wiley and Sons, 2011.
20. Y.-H. Liu, J. Deng, J.-W. Gao, Z. H. Zhang, Adv. Synth. Cat. 2012, 354, 441–447.
21. A. O. Terent'ev, M. M. Platonov, A. S. Kashin, G. I. Nikishin, Tetrahedron 2008, 64, 7944–7948.
22. K. B. Landenberger, O. Bolton, A. J. Matzger, Angew. Chem. 2013, 125, 6596–6599, Angew. Chem. Int. Ed. 2013, 52, 6468–6471.
23. E. Climent, D. Groeninger, M. Hecht, M. A. Walter, R. Martinez-Manez, M. G. Weller, F. Sancenon, P. Amoros, K. Rurack, Chem. Eur. J. 2013, 19, 4117–4122.
24. A. O. Terent'ev, M. M. Platonov, E. J. Sonneveld, R. Peschar, V. V. Chernyshev, Z. A. Starikova, G. I. Nikishin, J. Org. Chem. 2007, 72, 7237–7243.
25. A. O. Terent’ev, D. A. Borisov, I. A. Yaremenko, V. V. Chernyshev, G. I. Nikishin, J. Org. Chem. 2010, 75, 5065–5071.
26. A. O. Terent’ev, D. A. Borisov, V. V. Semenov, V. V.Chernyshev, V. M. Dembitsky, G. I. Nikishin, Synthesis, 2011, 13, 2091–2100.
27. A. O. Terent’ev, A. V. Kutkin, M. M. Platonov, Yu. N. Ogibin, G. I. Nikishin, Tetrahedron Lett. 2003, 44, 7359–7363.
28. D. V. Kazakov, O. B. Kazakova, G. Yu. Ishmuratov, A. O. Terent’ev, G. I. Nikishin, G. A. Tolstikov, Doklady Chemistry 2011, 436, 34–38.
29. A. O. Terent'ev, D. A. Borisov, Vera A. Vil’, V. M. Dembitsky, Beilstein J. Org. Chem. 2014, 10, 34–114.
30. A. O. Terent’ev, D. A. Borisov, V. V. Chernyshev, G. I. Nikishin, J. Org. Chem. 2009, 74, 3335–3340.
31. A. O. Terentev, I. A. Yaremenko, V. A. Vil’, I. K. Моisееv, S. A. Kon’kov, V. M. Dembitsky, D. O. Levitsky, G. I. Nikishin, Org. Biomol. Chem. 2013, 11, 2613–2623.
